Computational Analysis of Picosecond Protein Motions
|
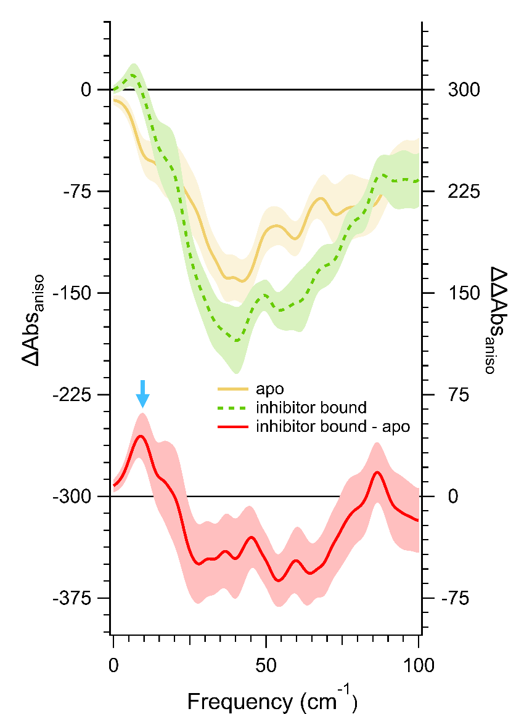
It is critical to complement our measurements with calculations to understand the results. First we need to calculate the specific dynamics that contribute to the measurement and then formulate those calculations into the measured quantity to directly compare to the experimental results. Ideally one would exactly model the measured system, for example, 1 mM protein solutions or protein crystals. These can be massive calculations. We simplify these calculations by doing single molecule calculations within a hydrated box with periodic boundary conditions. Our methods for determining the terahertz absorption then use either dipole autocorrelation or normal mode ensemble analysis. |
 |
Return to Research |